In the general molecular docking calculation, an indispensable step is to define the binding position of the ligand molecule (usually an organic small molecule), ie to the interface pocket. For the protein-small molecule complex X-ray crystal structure, there is a ligand in the pocket that indicates the position of the interface pocket for us. However, there are still many X-ray crystal structures and NMR-analyzed structures without ligand structure. How do we determine the interface bag? More generally, how do you define a pair of interface pockets for nucleic acids, peptides, and host molecules in the host and guest?
The concept of interface pockets exists in molecular docking calculations and is a possible region of ligand binding in the receptor. Tell the docking program its position by enclosing the pocket with a large enough box. If the interface pocket is set to a true active binding site, there is a greater probability of finding the correct active conformation and binding mode of the ligand. As for the interface bag, as the name implies, it is usually in the form of a pocket (small opening, large belly, capable of accommodating a certain volume of molecular structure), and other shapes, such as a tube shape, a groove shape and a shallow shape, and the shape of the pocket is most typical . For protein-ligand complexes, large and deep hydrophobic cavities are critical for ligand binding. For protein structure, this feature has become an important basis and principle for various algorithms to find interface pockets/identification binding sites.
Method for identifying protein pocket/ligand binding sitesThe following is a general strategy for determining the interface bag for the protein. Because the characteristics of pockets of nucleic acids, peptides, and host molecules vary widely, it is difficult to generalize, but if they are flexible, these strategies are often applied, and even easier to operate.
First, the literature / database research methodThe most reliable information is the experimental data. Through literature research, we can learn the main function and family of the protein from the experimental results of others/predecessors, and find the active site information . For example, the His-Asp-Ser of chymotrypsin catalyzes the Zn2+ binding site of the triplet and zinc finger protein. By comparing the homologous proteins of other species that have been studied more, the corresponding pocket information is found . The UniprotKB database (https://) integrates rich protein structure-function information, and some also contain mutation site information . This provides a reliable basis for our identification of protein pocket/ligand binding sites.
Second, the experimental screening methodIf there is a lack of useful information in the literature and database, and the experimental method is quite convenient and cheap, then do your own experiments, such as site-directed mutagenesis (usually used to verify the results of the calculations, not the opposite), fluorescent probe labeling [1] ].
Third, software prediction methodThis is the easiest and most direct way. There are a number of software/algorithms that can help people predict the active pocket of a protein or identify a binding site for a ligand. The figure below summarizes the basics of some of the forecasting procedures.

(Protein pocket/ligand binding site prediction local program or online service, click here to jump to the relevant link page)
The requirements of the input content of each program are similar, and the output results vary widely. Interested readers can explore. Here, the online service POCASA is used as an example to explain how to predict the pocket of the protein receptor 1UWH. The crystal structure is a protein-ligand complex, and the ligand molecule indicates the binding site, which is used to verify whether POCASA can correctly predict the pocket position.
1. Log in to the POCASA website: http://altair.sci.hokudai.ac.jp/g6/service/pocasa/
2. Enter 1uwh in the PDB ID field, keep the default values ​​for other parameters, and click the Get Pockets and Cavities button.
Wait a moment to return the result. The Chain ID defaults to NULL, which means that the first chain of the protein file is selected. The crystal structure of the protein contains both A and B chains, all of which have ligand small molecules. We predict the position of the pocket on the A chain.

(POCASA submits the task interface)
3. Find the Output files from the returned results and download the pdb file we need.
File 1 is the input pdb file (we entered the PDB ID, POCASA automatically downloads the protein file from the RCSB PDB library), and file 2 is the output we need, including the location information for several potential pockets. Download the two and observe the analysis using PyMOL or other molecular graphics software.

(POCASA output file, where XXXX_TopN_pockets.pdb is the predicted pocket location)
In addition, under the Rank order column, POCASA also tells us how many Pockets are generated. Each Pocket has its own number, sorted by volume, followed by Rank 1, 2, 3... Usually, the largest Pocket has the most It may be a real protein pocket, but it may be a fake pocket if it is too big. The safest approach is to perform a visual analysis.

(POCASA calculates the pocket size and pocket likelihood order)
4, using PyMOL visual analysis
Open the 1uwh.pdb and 1uwh_TopN_pockets.pdb files, hide the redundant structure, display the protein A chain in the form of cartoon, display the ligands in the form of sticks, and display the pockets in the form of spheres.

(The protein is a dark green strip, the ligand is a pink-orange stick, and the Pocket A~F is represented by a small ball of various colors)
It can be seen that the largest pocket is not exactly the binding pocket of the ligand; Pocket A only has about half of the volume overlaps with the ligand, Pocket B and the ligand molecule mostly overlap, and the two pockets together form the ligand binding pocket. As this example shows, we can't fully believe the results of software predictions, just look at the size of the volume, and may judge the mistakes. In practice, it should take time to examine the various prediction pockets.
Fourth, manual observationAccurate identification of protein pocket/ligand binding sites is inseparable from manual observation and analysis, and it is very dangerous to rush to conclusions only by software prediction . Using POCASA to predict the pocket position of the 1uwh protein, Pocket A and B form the binding pocket of the ligand, but there is a little trick inside. According to the "induction fit" theory, in the process of ligand binding, protein and ligand will undergo different degrees of conformational adjustment to achieve the "most comfortable" state. This state is different from free protein (protein without ligand binding). The above example uses a protein that is actually a complex for prediction, and the probability of success is greater. However, in practice, proteins that need to be predicted by the pocket are often ligand-free. Therefore, we cannot expect that the results of software predictions will always be as obvious as the examples (Pocket A and B are significantly larger than others). In some proteins with atypical pockets and multiple pockets, the software probably cannot predict a valid Pocket or predict multiple Pockets. This requires manual observation to eliminate Pockets with very low probability and pockets with high probability of retention .
Continued from the above example, using PyMOL to display the (van der Waals) molecular surface of the protein. By observation, we found that the two pockets each formed a sub-pocket, and the ligand molecules spanned both. Pocket A is large enough to form a large spatial area with Pocket F; Pocket B is more typical, deeper and narrower ; other Pockets are either too small or almost completely exposed to solution and are not suitable for use as pockets. Thus, both Pocket A and B have potential pockets. Without ligand molecules, we are not sure which one is the real pocket, or both are or not. This is a ubiquitous situation. It is rigorous to exclude (unlike the above criteria) the obvious Pocket, and leave the remaining pockets as candidate pockets for further elimination or identification in subsequent studies (such as molecular docking) .
Remember the principle of finding pockets given above? Yes, the binding of the ligand requires hydrophobic action. In general, hydrophobic cavities are more likely to become pockets. The feasibility of the pocket can be further judged by the hydrophobic distribution surface of the protein. Of course, the inside of the protein is usually hydrophobic, and it is also possible to roughly determine whether the hydrophobicity of a predicted pocket is sufficient from the shape and position . PyMOL is not very convenient to make a hydrophobic distribution surface, we will ignore this step. In addition, the docking score can also reflect the degree of hydrophobicity of the pocket to a certain extent, so that the protein pocket can be screened and the correct binding site can be identified .

(The light purple protein surface shows large and small cavities, and the predicted Pocket ball indicates the position of the potential pocket)
Define the interface bag on the Yin Fu cloud computing platform
Having said that, when using free protein as a receptor in molecular docking, how do you define the interface bag?
The computing platform provides us with three ways to define pockets. For complex proteins, you can define the ligand molecules that were extracted before by selecting the file ( see the platform tutorial, reply to the calculation tutorial on the WeChat public account home page). "You can get the download link "; for free proteins, you can define them by uploading a molecular file containing the pocket information or by selecting the amino acid residues in the pocket from the drop-down list.
Still taking 1UWH as an example, we put the protein at an angle similar to the above image, we know the approximate position of the pocket (green circle below), and then find one or several amino acid residues in the pocket (requires its atomic collection) The geometric center is as close as possible to the center of the pocket. Place the mouse over it and the relevant information will be displayed (yellow circle below). Then, check the residues in the drop-down list (the red box below).

(Defining the interface bag by specifying amino acid residues on the Yin Fu cloud computing platform)
Another convenient way is to upload a molecular file (using pdb, mol2, sdf, etc. [2-4]) to the platform in the center of the pocket, and the platform will calculate their geometric center to determine the center of the interface bag. position. For example, use the text editor NotePad++ to open the POCASA output file 1uwh_TopN_pockets.pdb, delete the Pocket C~F information, retain the information of Pocket A and B, save the pdb file, and upload it to the platform. POCASA assigns different chain names A~F to each Pocket very intimately; according to the Rank order information mentioned above, Pocket A has a residue name of 222 and Pocket B of 146. According to this, you can quickly find all the information of the two pockets.
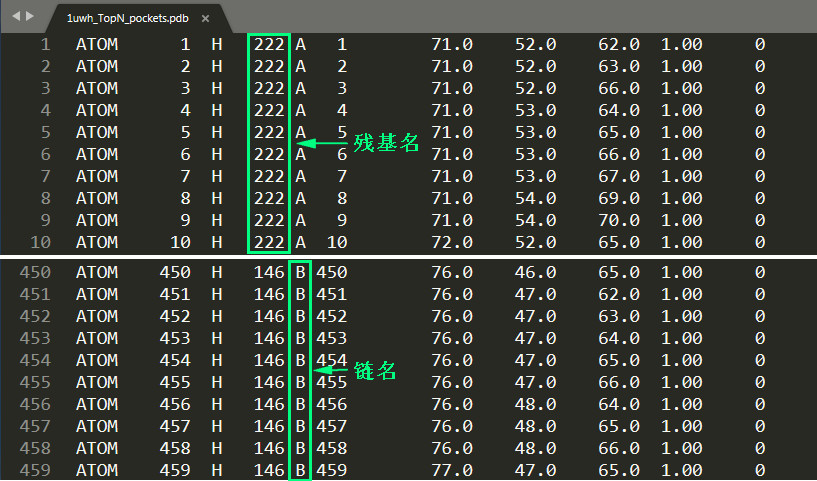
(Use a text editor to process the Pocket information in the POCASA output file)
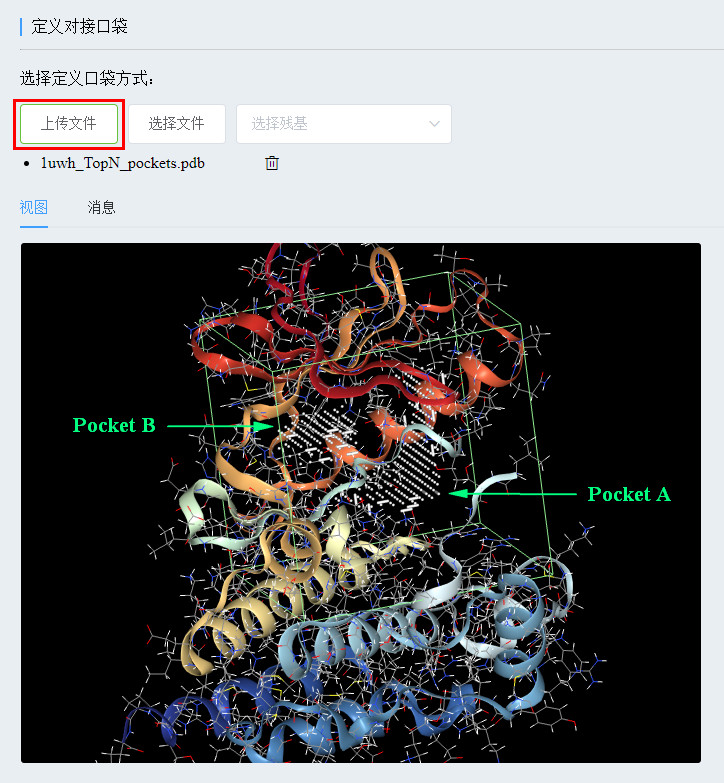
(Define the interface bag by uploading the deleted POCASA 1uwh_TopN_pockets.pdb file)
Of course, this method is not limited to the output file of POCASA. You can also use the output files of other predictive software. You can also upload your own created files. For example, you can select several amino acid residues in the pocket and save them as pdb files through software such as PyMOL. If the format is incorrect, the platform will throw an exception error. For users who don't know much about the molecular file format, it is recommended to use a drop-down list to define the interface bag.
[Next Announcement] We will release the 5-minute molecular docking video tutorial for the Vina and Dock6 programs in the next issue, so stay tuned.
references
1. Tina Seifert et al. Identification of the Binding Site of Chroman-4-one-Based Sirtuin 2-Selective Inhibitors using Photoaffinity Labeling in Combination with Tandem Mass Spectrometry. J. Med. Chem. 2016, 59, 23: 10794-99 DOI: 10.1021/acs.jmedchem.6b01117
2. PDB format: http://
Mol2 format: http://chemyang.ccnu.edu.cn/ccb/server/AIMMS/mol2.pdf or http:// .41.html
SDF format: http:// or http://link.fyicenter.com/out.php?ID =571
For more information, please visit or follow the WeChat public account "Yin Fu Technology". Our company has established the WeChat academic exchange group to build an interactive platform for communication and communication among friends in the biomedical field. If you want to join a group of friends, please enter "Add Group" in the WeChat public account menu bar and follow the prompts.